
Clinical Care Guidelines
- Introduction Clinical Care Guidelines
- Chapter 1 The Fanconi Anemia DNA Repair Pathway
- Chapter 2 Diagnosis of Fanconi Anemia: Testing and Genetic Counseling
- Chapter 3 Clinical Care of Fanconi Anemia Hematologic Issues
- Chapter 4 Non-HNSCC Solid Tumors in Patients with Fanconi Anemia
- Chapter 5 Head and Neck Cancer in Patients with Fanconi Anemia
- Chapter 6 Oral Health Care for Patients with Fanconi Anemia
- Chapter 7 Gynecologic Care for Female Patients with Fanconi Anemia
- Chapter 8 Dermatologic Issues in Patients with Fanconi Anemia
- Chapter 9 Clinical Care of Fanconi Anemia Gastrointestinal Issues
- Chapter 10 Endocrine Disorders in Patients with Fanconi Anemia
- Chapter 11 Hearing and Ear Issues in Patients with Fanconi Anemia
- Chapter 12 Clinical Care of Hand and Arm Abnormalities in Fanconi Anemia
- Chapter 13 Brief Guide to Clinical Care for Patients with Fanconi Anemia
- Appendix A: Glossary and List of Abbreviations
- List of Contributors
Chapter 2
DIAGNOSIS OF FANCONI ANEMIA:
TESTING AND GENETIC COUNSELING
Introduction
Fanconi anemia (FA) is a rare genetic disorder that results from DNA repair defects arising from pathogenic variants (PVs) in at least 22 genes (FANCA, FANCB, FANCC, FANCD1/BRCA2, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ/BRIP1, FANCL, FANCM, FANCN/PALB2, FANCO/RAD51C, FANCP/SLX4, FANCQ/ERCC4/XPF, FANCR/RAD51, FANCS/BRCA1, FANCT/UBE2T, FANCU/XRCC2, FANCV/REV7/MAD2L2, and FANCW/RFWD3) discovered to play a role in the FA DNA repair pathway (see Chapter 1). All PVs in these genes are inherited in an autosomal recessive manner except PVs in FANCB and FANCR/RAD51, which are X-linked and autosomal dominant, respectively. The carrier frequency of any PV in one of the FANC genes is 1:181 in the general population in the United States and 1:93 in Israel [1]. Specific populations may exhibit a founder effect, with increased PV carrier frequencies in certain FA-associated genes. Early diagnosis and characterization of patient-specific PVs are of utmost importance, as this information may affect a patient’s disease risks and clinical management, especially in severe cases. This chapter describes the importance of early diagnosis and the role of genetic counseling and cytogenetic and molecular tests used to diagnose FA. It also discusses test interpretation considerations to obtain an accurate diagnosis, which is important for guiding clinical management and facilitating appropriate testing of family members.
-Chapter updated in 2026-
Clinical Manifestations and Evaluation for Diagnosis
Most patients with FA have identifiable manifestations either at birth or during childhood [2]. The median age at diagnosis is 7 years [3, 4], although more severe phenotypes are typically diagnosed at a younger age [5]. Individuals with no obvious physical defects may not be diagnosed until adulthood unless they develop bone marrow failure (BMF) (see Chapter 3) or a solid tumor (see Chapters 4 and 5)[4, 6].
Physical Phenotype
Physical phenotypes associated with FA are multisystemic and extremely heterogenous, which can offer clues for testing and early diagnosis [4, 7]. The classical congenital abnormalities seen in FA are those described in the VACTERL-H (Vertebral, Anal, Cardiac, Tracheoesophageal fistula, Esophageal or duodenal atresia, Renal, upper Limb, and Hydrocephalus) association [8]. Approximately 5 to 30% of patients with FA meet the criteria for VACTERL-H association (≥3 of 8 features) [9, 10, 11]. Among the VACTERL-H features, the combination of renal and limb malformations is highly suggestive of FA [8]. Other abnormalities frequently seen in FA include those described by the acronym PHENOS (skin Pigmentation, small Head, small Eyes, Nervous system, Otology, and Short stature) [10]. The most frequent abnormalities described in retrospective analyses include short stature (43%), upper limb (radial ray) abnormalities (40%), skin pigmentation changes such as café au lait macules (37%), renal malformations (27%), microcephaly (27%), and male genitalia abnormalities (16%) [4, 9]. All these abnormalities, except those affecting only males, are included in VACTERL-H or PHENOS. Although most patients will have at least one abnormality, up to 40% (range: 10-40%) will have none; thus, the absence of abnormal features does not exclude the diagnosis of FA [9, 10, 11, 12, 13].
The information summarized in Table 1 can be used as a guide for evaluating patients whose appearance is suggestive of FA. Any combination of the abnormalities listed in Table 1 should raise the level of suspicion for FA.
Table 1. Manifestations that are indicators for Fanconi anemia screening.
| System | Clinical Feature |
|---|---|
| Growth | Intrauterine growth retardation; small for gestation age; short stature |
| Central Nervous System | Microcephaly; pituitary abnormalities; hydrocephalus; Bell’s palsy; central nervous system arterial malformations; absent septum pellucidum and/or corpus callosum; other anomalies of the corpus callosum; hyperreflexia neural tube defects; Arnold-Chiari malformation; Moyamoya; single ventricle hypoplastic adenohypophysis; platybasia; hypoplastic clivus; dilated ventricles |
| Ophthalmology | Microphthalmia; epicanthal folds; small palpebral fissures; telecanthus; short or almond-shaped fissures; microcornea; ptosis; strabismus; cataracts |
| Otology | Hearing loss or deafness; abnormal or absent pinna; low set or posteriorly rotated ears; small or absent ear canals; absent tympanic membrane; microtia; fused ossicles |
| Cardiopulmonary | Patent ductus arteriosus; atrial septal defect; ventricular septal defect; coarctation of the aorta; pulmonic or aortic stenosis; double aortic arch; cardiomyopathy; tetralogy of Fallot; pulmonary atresia; absent or displaced radial artery |
| Gastrointestinal | Tracheoesophageal fistula; esophageal, duodenal, or anal atresia; imperforate anus; annular pancreas; intestinal malrotation or obstruction; duodenal web; biliary atresia; foregut duplication cyst |
| Genitourinary | Urinary: horseshoe, sigmoid, ectopic, rotated, hypoplastic, dysplastic, or absent kidney; hydronephrosis; hydroureter; urethral stenosis; reflux Male reproductive: microphalus; penile and/or scrotal fusion; cryptorchidism; atrophic or absent testes; hypospadias; chordee; phimosis Female reproductive: hypoplastic, absent, or bicornuate uterus; aplasia or hypoplasia of the uterus; vagina atresia or agenesis; gonadal dysgenesis |
| Skeletal | Thumb and radial: thenar hypoplasia; absence or hypoplasia of radius and/or thumb; floating thumb; bifid thumb; digitalized thumb or abnormal thumb placement Craniofacial: triangular face; pointed chin; mid-face hypoplasia; microsomia; hypertelorism or hypotelorism; craniosynostosis; high arch or cleft palate; micrognathia; frontal bossing Other skeletal: dysplastic or absent ulna; polydactyly; clinodactyly; carpal bone hypoplasia; spina bifida; Klippel-Feil; vertebral anomalies; absent clavicles; Sprengel’s deformity; Perthes disease; congenital hip dysplasia or dislocation; scoliosis; rib abnormalities; clubfoot; sacral agenesis (hypoplasia); leg length discrepancy; kyphosis; brachydactyly; arachnodactyly; humeral abnormality |
| Integumentary | Café au lait macules; hypopigmentation or hyperpigmentation |
Sources for Table 1: [4, 6, 9, 14, 15, 16, 17, 18, 19, 20]
Diagnostic Testing
Any patient suspected of having FA should be referred to a hematologist and/or clinical geneticist or genetic counselor, who can arrange for diagnostic testing. As FA testing is highly specialized, particularly the evaluation of chromosome breakage in response to DNA damage, only laboratories with extensive experience should undertake this testing.
All laboratories (both cytogenetic and molecular) involved in testing should be accredited by a recognized regulatory body and certified to perform FA testing for clinical care. Recognized accreditation bodies in the United States, Canada, and Europe are as follows:
United States
- Clinical Laboratory Improvement Amendments and the College of American Pathologists provide laboratory certification and accreditation.
- The American College of Medical Genetics and Genomics (ACMG) provides detailed guidelines for cytogenetic testing and the interpretation of genetic testing results [21].
Canada
- The Ontario Laboratory Accreditation and the Canadian College of Medical Genetics provide laboratory oversight and guidelines, respectively.
Europe
- The Belgian Accreditation Council, French Accreditation Committee, Deutsche Akkreditierungsstelle, the Swiss Accreditation Service, and United Kingdom Accreditation Service provide accreditation services.
The recommended testing procedures are outlined in the flow chart in Figure 1. The flow chart presents one potential algorithm for testing, starting with chromosome breakage testing and ending with genotyping. However, genetic testing has become increasingly utilized as a first-line diagnostic test, especially for newborns and pediatric patients with multiple congenital anomalies. Regardless of the order of testing, it is important that both chromosome breakage and germline genetic testing be performed to obtain a precise diagnosis in all persons with suspected FA.
Chromosome Breakage Test in Peripheral Blood Lymphocytes
The chromosome breakage test is the first test that should be performed for an individual suspected of having FA. This assay is performed in a clinical cytogenetics laboratory, often using a sample of the patient’s peripheral blood. Peripheral blood is treated with diepoxybutane (DEB), a DNA crosslinking agent. Mitomycin C (MMC) is another crosslinking agent commonly used for this assay, although it may lead to a higher rate of false positives than DEB [22, 23]. Following exposure of peripheral blood cells to these DNA damaging agents, the chromosomes are examined for evidence of chromosomal damage [6, 24]. Cells from individuals without FA will have relatively few chromosome breaks or rearrangements. In contrast, cells from patients with FA will exhibit multiple chromosome breaks and rearrangements per cell, including complex rearrangements such as radial figures, which are the hallmark abnormality of this disease. As recommended in the ACMG guidelines for cytogenetic laboratories [25], the test results report should include the following: (1) breakage and rearrangement rates, (2) distribution of chromosome breakage among cells, (3) average number of aberrations per cell, and (4) percentage of cells with radials.
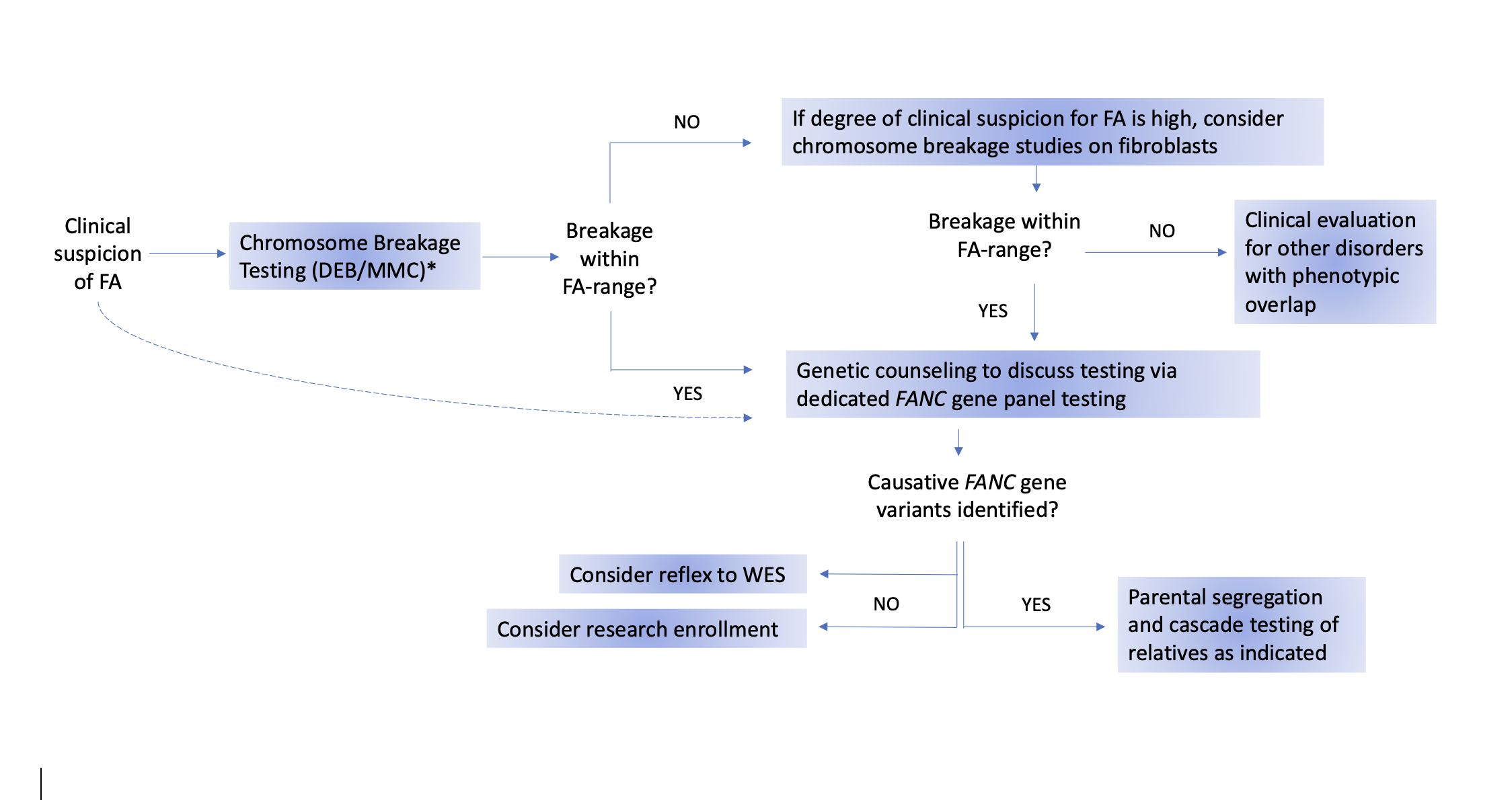
Figure 1. Suggested algorithm for Fanconi anemia testing.
The gold-standard test for diagnosing Fanconi anemia (FA) is the chromosome breakage test using DNA crosslinking agents, specifically diepoxybutane (DEB). Mitomycin C (MMC) may also be used, but it has a higher potential for false-positive results [22, 23]. Dotted line: If chromosome breakage testing is unavailable, a diagnosis of FA can be directly confirmed by genetic testing. If peripheral blood testing is negative but clinical suspicion for FA remains high, the negative result may be a false negative because of somatic mosaicism in the peripheral blood. Therefore, repeat testing on fibroblasts cultured from a skin biopsy should be performed. If the chromosome breakage testing is positive, FA panel testing should be performed. If FA panel testing is negative, whole exome sequencing (WES) and/or research testing should be considered. In cases where chromosome breakage testing on both blood and skin cells is negative, referral for evaluation of conditions that overlap clinically with FA should be considered.
The laboratory should also measure baseline chromosome breakage by evaluating cells that have not been treated with DEB and/or MMC when performing the chromosome breakage test. The measurements of baseline breakage can vary markedly among patients with different FA variants. For example, patients with variants in the FANCD1/BRCA2 orFANCN/PALB2 genes have elevated levels of baseline breakage and unusual constellations of abnormalities (balanced translocations, derivative chromosomes, inversion, and rings), compared with other genotypes [26]. Assessment of baseline breakage may also aid in diagnosing other chromosome instability disorders that display specific types of chromosomal abnormalities. These abnormalities include the following: (1) rearrangements of chromosomes 7 and/or 14, which commonly occur in ataxia-telangiectasia and Nijmegen breakage syndrome; (2) telomeric rearrangements and dicentric chromosomes, which are often found in dyskeratosis congenita; and (3) premature centromere separation, which is characteristic of Roberts syndrome and Warsaw breakage syndrome [27, 28, 29, 30, 31, 32, 33].
Interpreting Chromosome Breakage Test Results
Although the chromosome breakage test is considered the gold standard for diagnosing FA, it may still produce false-negative results (the test is negative, but the patient has FA) or false-positive results (the test is positive, but the patient does not have FA). The following sections describe situations when false positives or negatives may occur.
Positive Test Result
Positive chromosome breakage test results considered diagnostic for FA must demonstrate DEB- or MMC-induced chromosomal aberrations. Chromosome analysis must be performed in metaphases with solid-color staining (without chromosomal banding), which limits visualization of chromosome aberrations [34]. The evaluation should include scoring of 50 metaphases in each culture. If more than 30% of cells have radials, only 25 metaphases are required for scoring. For each metaphase, the number and type of chromosomal aberrations should be evaluated, with the aberrations grouped as follows: chromatid and chromosomal breaks, centric and acentric fragments, rings, dicentrics, radial figures, and others [34].
The presence of radial figures is a decisive factor in determining the diagnosis of FA. Radial formation caused by crosslinking agents is considered the hallmark of the disease and distinguishes FA cytogenetically from other chromosomal breakage syndromes. Fanconi anemia is diagnosed when there is an increased percentage of cells with radial formations. Other types of aberrations, such as breaks, dicentrics, and fragments, are nonspecific [35, 36]. Therefore, the number of aberrant cells, the degree of damage in the aberrant cells, and the types of aberrations are taken into consideration for the final diagnostic determination. The test is considered positive for FA when the percentage of cells with radials is 30% or higher in DEB culture [37].
After a positive chromosome breakage test result, a hematologist or genetic counselor can help coordinate the necessary follow-up. Importantly, follow-up testing should be performed to identify the patient’s PV using the genotyping methods described later in this chapter.
In some cases, a diagnosis of FA is suspected only after an individual is diagnosed with cancer, such as leukemia or a solid tumor. The physician may suspect FA when a patient experiences severe adverse effects from genotoxic therapies used to treat the malignancy. In these patients, a DEB or MMC chromosome breakage test of peripheral blood is warranted, but interpretation may be confounded by the elevated baseline chromosomal breakage that may be observed in blood samples from patients treated with chemotherapy or radiation [23]. In such cases, testing cultured skin fibroblasts may be helpful.
Negative Test Result
A chromosome breakage test result is considered negative if metaphase cells from DEB- or MMC-treated cultures do not show increased chromosomal radial formation or if the rate of observed breakage or radials falls within the laboratory’s established normal range. If the chromosome breakage test is negative and clinical evidence for FA is weak, no further studies are required. However, if there is strong clinical evidence for FA, skin fibroblast testing should be performed to rule out the possibility of somatic mosaicism. In addition, there are multiple disorders with clinical features common to FA that are associated with some form of chromosome instability that could be considered [27, 28, 29, 30, 31, 32]. If the chromosome breakage test is negative in a patient with multiple, unexplained clinical features, the patient should be referred for additional genetic evaluation.
Equivocal Test Result
Test results are considered equivocal, or inconclusive, if the percentage of cells that display chromosomal breakage patterns characteristic of FA is lower than the laboratory threshold for FA or if there is increased breakage, but the types of breakage are not characteristic of FA. The percentage of cells with radials may fall above the upper limit of the normal control range but below the lower limit of the laboratory’s FA range. Underlying causes of inconclusive results include mosaicism in the patient’s peripheral blood cells, hypomorphic variants in FANC genes, and the presence of a condition other than FA that manifests with increased chromosomal breakage.
Mosaicism in Peripheral Blood Cells
Somatic mosaicism can occur in the T-lymphocytes and hematopoietic stem cells of individuals with FA by various mechanisms. Additional testing of cultured fibroblasts from a skin biopsy should be performed if clinical evidence for FA is strong, but peripheral blood chromosome breakage test results are negative or equivocal. The diagnosis of FA can be confirmed by a chromosome breakage test that reveals increased breakage in fibroblasts, with the types of breaks and rearrangements characteristic of FA (radial formation). Some degree of mosaicism is present in 15 to 20% of patients with FA, where their fibroblast cultures show increased breakage, while the lymphocytes do not. Pathogenic variant reversion in hematopoietic stem cells has been documented in less than 5% of patients with FA [38]. An initially low percentage of cells with somatic reversion (no longer characterized as FA cells) may increase over time and may be accompanied by spontaneous improvement in blood cell counts. However, mosaicism measured in peripheral blood lymphocytes may not reflect mosaicism in hematopoietic stem or progenitor cells. Therefore, patients with a high percentage of reverted cells in the tested lymphocytes may have no (or a very low percentage of) reverted cells in their bone marrow. As bone marrow cells are involved in the development of leukemia, their status should not be generalized from peripheral blood lymphocyte results; thus, it remains unclear whether the clinical disease course will be altered in patients with reverted peripheral blood cells. Importantly, the presence of mosaicism—in either the blood or bone marrow—does not protect the individual from developing clonal chromosomal abnormalities within the population of cells that retain their FA phenotype, which may lead to the development of hematologic malignancies. Mosaicism in the blood or bone marrow also does not protect against the development of solid tumors.
Germline Genetic Testing
If the results of the chromosome breakage test are positive, genetic testing should be performed to identify the specific PV(s) associated with the patient’s FA phenotype. Genetic testing enables accurate diagnosis, which may improve clinical care for individuals with anticipated genotype-phenotype associations and for relatives who are heterozygous carriers of PVs that confer an increased risk for malignancy (see Carrier Cancer Risk section in this chapter). Further, genetic testing is useful for preconception screening and prenatal diagnosis and is required for preimplantation genetic diagnosis (see Chapter 7).
Next Generation Sequencing
Until recently, a genetic test known as complementation analysis was the primary method available for determining which FANC genes were altered in patients with FA. However, complementation analysis is labor intensive, expensive, and time consuming. Over the past decade, the development and expansion of next generation sequencing (NGS) technologies (also referred to as massively parallel sequencing or multiplex testing) have transformed the field of genetic testing, as they enable detailed analysis of numerous genes simultaneously. Following a positive chromosome breakage test, NGS panel testing for clinically relevant FANC genes should be offered as the next step.
Clinical laboratories have evolved to offer two types of panel tests: dedicated panels (laboratory pre-selected genes associated with a patient’s phenotype) and custom panels (self-selection of relevant genes from a large list of genes). When selecting a panel, it is important to consider whether the test has been designed to address variant hotspots and/or gene regions known to present reporting challenges. As an example, the FANCD2 gene has two pseudogenes that can complicate the accuracy and interpretation of test results [39, 40]. Because of rapidly evolving knowledge of FA, many laboratories have not yet added the more recently discovered FANC genes to their panels. Thus, most currently available panels evaluate only a subset of the 22 known FANC genes [41, 42].
In addition to sequencing, testing should always include copy number analysis, which will allow the identification of large deletions, duplications, and insertions [43]. This is critical since 35% of patients with FA harbor large deletions that account for 18% of all FA PVs [44]. Because of the high rate of copy number variants (CNV), techniques that can detect gene deletions, duplications, and insertions, such as array comparative genomic hybridization, multiplex ligation-dependent probe amplification, and NGS-based copy number analysis, are an important part of the genetic testing process [45, 46, 47]. Detection of CNV can be performed in tandem with panel testing or as a reflex test. When clinical evidence of FA is less convincing, broader panels targeting a specific phenotype, such as BMF, myelodysplastic syndrome (MDS), or acute myeloid leukemia (AML), may be considered. Broad panels are often not comprehensive for each of the syndromes they analyze, so an FA-specific panel is still preferred when the diagnosis of FA is considered likely.
Whole Exome and Whole Genome Sequencing
Whole exome sequencing (WES) is an NGS approach that is more expansive than sequencing targeted panels of genesand aims to sequence all exons and splice sites of all known genes, which represent approximately 2% of the human genome. An even more expansive NGS application is whole genome sequencing (WGS), which analyzes the entire human genome, and has recently become more widely available for clinical use; however, the analysis remains mostly focused on exons and splice sites, and the ability to interpret the effects of variants outside of these regions remains limited. The high cost of WES and WGS currently prohibits their use as first-line testing tools. Whole exome sequencing may be warranted for individuals with a diagnosis of FA based on a positive chromosome breakage test but with no known causative variants identified on dedicated FA panel testing.
Targeted panels can identify novel PVs within known FANC genes, but only tests such as WES or WGS can identify novel FANC genes because these latter tests screen specific regions of the genome and beyond [40]. Additionally, WGS analyzes regions within known FANC genes, such as deep intronic or promoter variants, which may not be covered by other methods. Therefore, WGS may detect novel PVs in classic FANC genes. While WES and WGS can detect variants in larger areas of the genome than panel testing, these NGS methods have risks and limitations (see Table 2). Both WES and WGS may identify a greater number of variants of uncertain significance (VUS) in FANC or other genes and may create ethical dilemmas if abnormal findings unrelated to the patient’s phenotype are detected [40, 48]. These possibilities should be presented to the patient and/or family in advance. Genetic counselors and geneticists are experienced in conducting informed consent conversations and ordering broad sequencing tests like WES or WGS. They can assist with interpretation of the results and should be involved in discussions of the results with providers and families. Table 2 provides an overview of the benefits and limitations of sequencing using dedicated gene panels, WES, or WGS.
Table 2. Benefits and limitations of current next generation sequencing platforms.
| Platform | Benefits | Risks or Limitations |
|---|---|---|
| Dedicated gene panel | • Clinically available genes associated with a patient’s phenotype are analyzed in a single test. • Certain regions may be specifically addressed to capture accurate data on known recurrent PVs and sequencing challenges (e.g., FANCD2 pseudogenes). • Fast turnaround time and lowest cost option. |
• Will not detect larger deletions or duplications if copy number analysis is not included. • May fail to detect deep intronic variants or variants in gene promoter regions. • Variants of unknown significance may be identified. • Incidental discovery of a hereditary cancer risk not associated with the underlying FA diagnosis is possible. |
| Whole exome sequencing (WES) | • All coding regions (exons) of the genome are sequenced in a single test. • May provide value for patients with no PVs identified by dedicated panel testing (may provide opportunity for gene discovery through research). • Can provide information for conditions other than FA if the diagnosis is uncertain. |
• Cannot detect larger deletions or duplications or structural changes, such as translocations and inversions. • Cannot distinguish pseudogene regions or detect deep intronic variants. • Overall coverage is poorer than with WGS, and some exons are not analyzed effectively. • May uncover findings unrelated to the patient’s diagnosis, with the potential for a greater number of VUS than panel testing. • More expensive and a slower turnaround time than panel testing. |
| Whole genome sequencing (WGS) | • All coding and non-coding regions (exons, introns, and regulatory regions) of the genome are sequenced in a single test. • May provide value for patients with no PVs identified on dedicated panel testing or WES (may provide opportunity for gene discovery through research). • Can provide information for conditions other than FA if the diagnosis is uncertain. |
• Standards of what defines a clinical genome are still emerging. • Assay cost, turnaround time, and variant interpretation require further refinement for WGS to be clinically relevant. • May uncover findings unrelated to the patient’s diagnosis, with the potential for a greater number of VUS than panel testing and WES. |
Special Considerations with Genetic Testing
Genetic Discrimination
Fear of discrimination is a common concern for patients when considering genetic testing. Genetic discrimination occurs when people are treated differently because they have a genetic variant that increases the risk of an inherited condition. The Genetic Information Nondiscrimination Act (GINA) is a United States federal law designed to protect people from health insurance and employment discrimination [49]. The GINA does not protect against genetic discrimination for other forms of insurance, such as life, disability, or long-term care insurance.
Variant Interpretation
Detection of VUS presents a major challenge when interpreting genetic testing results. As their name suggests, VUS are variations in genetic sequences for which no clear association with a phenotype has been established. Although healthy variation in the human genome is expected, the likelihood of finding sequence variations that are novel and difficult to interpret increases as the extent of the analyzed genome increases. The ACMG has recommended the use of a standard classification system to create a common language for clinical variant interpretation [21]. Based on specific criteria, a sequence change may be characterized in terms of its relationship to disease as pathogenic, likely pathogenic, VUS, likely benign, or benign. While pathogenic and likely pathogenic results are often sufficient to provide a genetic diagnosis, VUS findings should be interpreted with caution. If a VUS is found, families should be encouraged to remain in annual contact with their genetics team for updates on the interpretation of their specific variant(s) and to enter research studies assessing pathogenicity.
Variant Phasing
Although sequencing platforms can detect the presence of genomic variants, they may or may not detect “phase.” Phase refers to the genetic relationship between a pair of variants affecting the same gene, with variants classified as in cis or in trans. In cis variants are located in the same copy of the gene (both variants are on the same allele), whereas in transvariants are located on different copies of the gene (one variant is on one allele, and the other variant is on the opposite allele). Thus, to establish the diagnosis of autosomal recessive FA when two different PVs are found, parental testing should be offered to confirm that the variants are positioned in trans.
Secondary Findings
As the number of genes analyzed increases, so does the potential to identify additional findings that may or may not be related to the reason for testing. In addition to identifying the underlying genetic cause of an individual’s FA, larger panels, WES, and WGS may also reveal a variant in a gene linked to other health risks. In this scenario, the unanticipated variant is called a secondary finding. For example, testing may detect two FANCA gene variants that explain the patient’s FA phenotype and also identify a single PV in FANCD1 associated with hereditary breast and ovarian cancer syndrome [50]. Patients (and/or their parents or guardians in the case of children) should be told of this potential secondary finding in advance. In the case of WES and WGS, the ACMG has compiled a specific list of genes for which reporting of secondary findings is recommended [51]. As a critical component of a patient’s right “not to know,” it is important to review the opportunity to opt out of receiving secondary findings during the informed consent discussion for WES or WGS.
Negative Molecular Test Results
Negative molecular test results should be interpreted carefully in patients with chromosome breakage test results within the FA range. Possible explanations for a negative molecular test result include the following: (1) the presence of PVs in gene(s) that are not evaluated by the testing approach (e.g., a limited panel), (2) a type of variant in a gene that cannot be identified with current technology, (3) PVs in an undiscovered FANC gene, and (4) mosaicism [52, 53]. Analysis of an alternative type of sample (such as fibroblasts) may be considered in individuals presenting with an FA phenotype and negative genetic studies on peripheral blood if mosaicism is suspected.
Bone Marrow Analysis for Somatic Genetic Variation
Chromosome G-Banding Analysis
Following the diagnosis of FA, cytogenetic analysis of chromosomes in bone marrow cells should be performed using standard G-banding methodology. The goal of this analysis is to investigate for the presence of a clone with acquired chromosome abnormalities and, if present, to characterize the observed abnormalities. Identification of a clone, which involves the presence of the same numerical and/or structural chromosomal abnormalities in multiple cells, is an indication of an abnormal hematologic process. The significance of cytogenetic findings must be interpreted within the context of the clinical findings, bone marrow morphologic findings from hematopathology examination, and immunophenotyping results. It is also important to note that the cells of patients with FA demonstrate chromosomal instability, and it is therefore likely that some cells will develop random, non-clonal abnormalities. The clinical laboratory performing the chromosome analysis should have expertise in cancer cytogenetics, be familiar with FA and the types of abnormalities associated with the disorder, and be able to distinguish non-clonal abnormalities (which are limited to single cells and do not represent an emerging malignant process) from clonal abnormalities (which can herald the development of a premalignant or malignant condition).
Clonal Abnormalities
Myelodysplastic syndrome (MDS), acute myeloid leukemia (AML), and other hematologic malignancies are associated with clonal abnormalities; therefore, the observation of a clonal abnormality may presage the emergence of neoplasms or of a precancerous condition. Some clonal abnormalities in patients with FA may persist for a long time without adverse consequences, whereas others are associated with more rapid progression or more aggressive disease. In either situation, clonal evolution and clonal expansion are frequently associated with disease progression. If no clonal abnormalities are observed in the bone marrow, G-banding analysis should be repeated annually. If an abnormal clone is detected, follow-up analyses should be performed more frequently than once per year to monitor the behavior of the clone to evaluate for evolution or expansion. To allow comprehensive interpretation of the bone marrow chromosome analysis results, a hematopathologist should provide morphologic evaluation and flow cytometry with immunophenotyping to further characterize the abnormal cells.
Recurring clonal chromosome abnormalities may be found in patients with MDS, AML, and other cancers (see Chapter 3). Certain chromosomal abnormalities occur more frequently in patients with FA, including gain of material from the long arm of chromosome 1, gain of material from the long arm of chromosome 3, and loss of chromosome 7. These abnormalities can occur alone, or they may be seen in combination with each other or with other abnormalities involving other chromosomes [54, 55, 56, 57, 58]. One study found that gain of material from the long arms of chromosomes 1 and 3, and loss of chromosome 7 accounted for 75% of clonal abnormalities observed in patients with FA [54]. Gain of 3q material, in particular, is specific to FA and is frequently associated with cytogenetic evolution that includes monosomy 7 and leads to MDS. Accordingly, discovery of a 3q gain in a patient with apparent de novo MDS or AML should trigger a recommendation for DEB chromosome breakage testing.
Fluorescence In Situ Hybridization
Because a given clonal abnormality, such as gain of material on the long arm of chromosome 3, is often embedded within a more complex structural abnormality (e.g., a very small amount of material from 3q may be translocated to another chromosome), it may be difficult to accurately characterize the abnormality using G-banding alone. In such instances, fluorescence in situ hybridization (FISH), which employs fluorescently labeled chromosome region- or gene-specific probes, can be a highly informative addition to G-banding chromosome analysis. Other subtle abnormalities may be completely overlooked without performing FISH. While G-banding examines all chromosomes for abnormalities, FISH analysis typically examines cells for a small set of pre-specified abnormalities. Furthermore, G-banding is limited to dividing cells and is rather labor intensive, which limits the overall number of cells analyzed. In contrast, FISH analysis can be used to quickly examine more than 100 cells through interphase hybridization. Thus, these two techniques—G-banding and FISH—complement each other. Because gain of 1q and/or 3q and loss of 7 comprise the majority of clonal abnormalities in patients with FA, it is recommended that, in addition to the G-band analysis of 20 metaphase cells, FISH analysis of 100 to 200 interphase cells be performed to detect the presence of a small clone harboring one of these three abnormalities. Some laboratories use FISH analysis for a larger number of regions involved in MDS and AML (e.g., 5q, 20q) in patients with and without FA. These FISH panels can be applied to either unstimulated peripheral blood or to bone marrow specimens. The concordance of FISH results between blood and bone marrow samples in patients with FA has not yet been clearly established; however, some physicians and laboratories have begun to perform FISH analyses on peripheral blood samples collected at specific time points between annually scheduled bone marrow testing. This strategy of intervening blood FISH analyses is being evaluated as a noninvasive means of more frequently monitoring for the emergence of an abnormal clone with 1q or 3q gains or monosomy 7.
Genomic Microarray Testing
Genomic microarray testing is a relatively recent technique that has become a major tool for cytogenetics and molecular laboratories. Microarray techniques, such as array comparative hybridization and/or single nucleotide polymorphism analysis, can identify regions of chromosomal loss and/or gain that may be too small or too ambiguous in banding patterns or too complex to be identified by conventional chromosomal banding techniques. Sometimes, there are so many abnormalities in a single cell that a specific abnormality is essentially hidden. Microarray techniques are highly sensitive for detecting and identifying the origin of regions of chromosome loss and gain. For example, microarray techniques can rapidly detect and characterize the presence of a 3q abnormality and provide specific information about the boundaries of the gain region. However, one limitation of these techniques is that the clonal abnormality must be present in a sufficiently high percentage of cells (generally >10%) to be detected. Unlike FISH and conventional G-banding analyses, microarray analysis does not provide information about individual cells but rather provides results based on the total population of sampled cells. Nevertheless, given the now widespread availability of microarray testing, microarray analysis is recommended in cases with complex bone marrow chromosome results.
Genotype-Phenotype Associations in Fanconi Anemia
Fanconi anemia is a genetically and clinically heterogeneous disease. In some instances, knowing the mutated gene and specific PVs can be critical for identifying potential risks and attempting to understand the clinical disease course. While certain genotypes are associated with additional disease risks that necessitate risk screening beyond the classic FA management recommendations, other genotypes have not been associated with the classic features of FA. It is important to recognize that genotype-phenotype information is often based on a limited number of patients and that outliers to the traditional phenotypes have been observed. Several FA genotype-phenotype associations for which sufficient information is available are included in the following sections. For more information on associations between genotypes and physical phenotypes in FA, see Fiesco-Roa et al [9].
FANCA
One study reported that individuals with homozygous null PVs in the FANCA gene develop anemia at an earlier age and have a higher incidence of leukemia than individuals with residual function FANCA variants [59]. However, a separate analysis revealed that the age of onset of anemia and incidence of leukemia were not altered in patients with homozygous null FANCA PVs or in patients who express an abnormal form of the protein (hypomorphic PV) [60]. A recent study from Israel demonstrated that patients with FANCA PVs develop cancer at a significantly older age than those with other genotypes, but overall survival is not modified by the affected gene [61]. Specific variants in FANCA, such as the p.His913Pro and p.Arg951Gln/Trp variants, have been associated with later-onset disease and slow hematologic progression [62].
FANCB
Males with a truncating PV in the FANCB gene frequently present with overt findings consistent with VACTERL-H [9, 45, 63, 64]. A milder phenotype has been reported in patients with missense variants or somatic mosaicism [65, 66]. FemaleFANCB carriers (heterozygous) do not appear to exhibit associated disease findings [63].
FANCC
The International Fanconi Anemia Registry (IFAR) noted that individuals with PVs in FANCC had an earlier age of onset of BMF and poorer survival, compared with individuals with variants in FANCA or FANCG [67]. This finding was not reported by the European Fanconi Anemia Research Group, which described a less severe hematologic course and fewer somatic abnormalities in the FANCC group, compared with patients with FANCA and FANCG mutations [59]. Some PVs in the FANCC gene have been associated with specific phenotypes. Variants located in a region of the gene known as exon 15 (historically, exon 14) are associated with the development of blood abnormalities at an earlier age, more congenital abnormalities, and poorer survival, compared with variants in exon 2 (historically, exon 1) [67, 68]. The variant c.456+4A>T (previously known as IVS4+4A>T) was also reported to be associated with a more severe disease presentation in Ashkenazi Jewish individuals [68, 69]. However, this variant has been reported in other populations [70, 71] and may not be associated with a severe phenotype in some other groups [72]. Several studies suggested that the c.67delG founder variant (formerly known as c.322delG) is associated with milder symptoms, but exceptions have been reported [68, 69, 73]. A study of a Saudi population reported that the founder variant c.165+1G>T may also be associated with milder disease [74].
FANCD1/BRCA2
A study published in 2002 reported that individuals with FA and biallelic PVs in the FANCD1/BRCA2 gene may develop leukemia—especially AML or acute lymphoblastic leukemia (ALL)—at a much earlier age than those with other FA genotypes [75]. They are also at risk of developing solid tumors of the brain (e.g., medulloblastoma, glioblastoma multiforme, astrocytoma) and kidney (e.g., Wilms tumor), which are not typically seen in patients with FA [76, 77]. If a patient has biallelic FANCD1/BRCA1 PVs, additional screening with brain magnetic resonance imaging and kidney ultrasound should be considered [78]. A recent comprehensive review analyzing 71 patients with biallelic PVs in FANCD1/BRCA2 confirmed the association between these types of tumors and the FANCD1/BRCA2 genotype and showed that patients with the FANCD1 genotype can have multiple cancers and an earlier age of tumor presentation [79].
While some studies in patients with biallelic FANCD1/BRCA2 PVs have demonstrated a severe phenotype, including multiple congenital abnormalities and a 97% risk of developing any malignancy by 5.2 years of age [76], there are reports of older individuals with milder or later-onset disease and, in some cases, non-classic FANCD1/BRCA2-associated malignancies (e.g., lymphoma, colon adenocarcinoma, breast cancer) in early adulthood [75, 80, 81, 82, 83]. Additional reports of primary ovarian insufficiency (POI) in individuals with FA secondary to biallelic FANCD1/BRCA2PVs further suggest the possibility of a wide spectrum of disease in these patients [84, 85].
FANCG/XRCC9
The European Fanconi Anemia Research Group reported that individuals with PVs in FANCG/XRCC9 have more severe cytopenia and a higher incidence of leukemia than patients with variants in other FANC genes [59]. However, this pattern was not observed in the dataset analyzed by IFAR [67].
FANCM
Overall, more information is required to better understand the FANCM phenotype. FANCM was proposed in 2005 to operate as an FA core complex gene and was associated with an FA phenotype in a family with affected siblings [86]. Biallelic FANCA PVs were later identified in the affected siblings, raising the question of FANCM as a canonical FANCgene [87]. Biallelic loss of function FANCM variants have since been identified in individuals with a diverse range of phenotypes, including early-onset cancer (squamous cell carcinoma and breast cancer) with treatment toxicity without overt BMF or congenital abnormalities, chromosome fragility with congenital abnormalities and BMF, or fertility abnormalities (e.g., POI, oligospermia, azoospermia) [88, 89, 90, 91, 92, 93, 94].
FANCN/PALB2
Pathogenic variants in the FANCN/PALB2 gene are typically associated with a more severe clinical presentation. Similar to the FANCD1/BRCA2 phenotype, individuals with PVs in FANCN/PALB2 develop leukemia and a specific spectrum of solid tumors at an earlier age than patients with PVs in other FANC genes [79, 95]. Commonly reported tumors include medulloblastoma, Wilms tumor, AML, and neuroblastoma [79, 95, 96, 97]. In the absence of specific consensus guidelines, the cancer surveillance recommendations for patients with biallelic FANCD1/BRCA2 PVs also may be considered for individuals with FANCN/PALB2 PVs. Phenotypes outside this spectrum have been reported [98], indicating that additional cases may further expand the phenotypic spectrum of FANCN/PALB2-associated FA.
FANCO/RAD51C
Two families with an FA-like disorder and biallelic PVs in FANCO/RAD51C have been reported [99, 100]. In both families, the affected individuals presented with significant congenital anomalies, including some that are atypical for FA, such as palate anomalies, holoprosencephaly, and overlapping fingers. Hypersensitivity to DEB and MMC and increased radial breaks confirmed the diagnosis of FA. The risk of BMF and solid tumors remains unknown in patients with FANCO/RAD51C PVs.
FANCR/RAD51
While nearly all FANC genes require a PV on both gene copies (biallelic) to cause disease, only a single PV (monoallelic) is necessary in the FANCR/RAD51 gene. In two reported cases, de novo monoallelic PVs in FANCR/RAD51 were identified in the probands, resulting in an FA-like phenotype with congenital abnormalities but no apparent hematologic disease or cancer [101, 102].
Monoallelic PVs in FANCR/RAD51 have also been associated with congenital mirror movements, which is an autosomal dominant disorder with incomplete penetrance [103, 104, 105, 106]. To date, FA and congenital mirror movement phenotypes have not been described in the same individual.
FANCQ/ERCC4/XPF
In addition to the FA phenotype, biallelic PVs in FANCQ/ERCC4/XPF have been linked to autosomal recessive Cockayne syndrome, xeroderma pigmentosum, and a single case of XFE progeroid syndrome [107, 108, 109, 110]. Affected individuals can present with a single phenotype or concomitant phenotypes, depending on how gene function is impacted [107, 108, 109, 110].
FANCS/BRCA1
The first confirmed case of a biallelic FANCS/BRCA1 PV was reported in a 28-year-old woman with stage IV papillary serous ovarian carcinoma and severe toxicity to cisplatin treatment, although the diagnosis was not confirmed by chromosome breakage testing [111]. Collectively, nine cases have been described, with microcephaly, growth failure, skin hypopigmentation or hyperpigmentation, and facial dysmorphism observed in all cases. Five individuals presented with cancer between 2 and 30 years of age, including two patients with breast cancer and one each with ovarian cancer, T-cell ALL, and neuroblastoma; they exhibited varying degrees of sensitivity to chemotherapy and variable responses to interstrand crosslinking agents. None of these individuals presented with BMF or immunodeficiency [112, 113, 114, 115, 116].
Additional Genetic Counseling Considerations
The decision to proceed with any type of genetic analysis should be at the discretion of the patient or guardian. Genetic testing has benefits, risks, and limitations, which should be reviewed in advance so that an informed decision about testing can be made. The complexities of genetic testing necessitate a detailed conversation with a genetics counselor or geneticist, as misdiagnosis or misinterpretation of test results can have a significant impact on individuals and their family members. Patients should be counseled by an experienced genetics provider at the time of diagnosis and at various points throughout their lives. A genetics consultation should include discussions of the following:
- The genetic testing process
- Family, medical, and pregnancy histories
- Inheritance of FA
- Evaluation and follow-up by other specialists
- Reproductive options for patients, as well as their parents and other relatives
- Research opportunities
- Community support and resources
Family History
A detailed family history that encompasses at least three generations should be obtained for patients with FA. Family history can aid in identifying other family members with FA-related clinical features and in determining the inheritance pattern. Physical abnormalities, cytopenias, recurrent pregnancy loss, ancestry, and any family history of cancer should be noted.
Ancestry
Most PVs occur regardless of ancestral background. However, some PVs, referred to as “founder mutations,” are carried at an increased frequency in certain groups. Information about founder PVs can be useful for a few reasons, including defining the anticipated phenotype and estimating population-specific carrier frequency. Examples of both scenarios are as follows:
- Defining the anticipated phenotype:
- The FANCC c.67delG PV, which is common in Northern Europeans [68], and the FANCA p.His913Pro PV, which appears to be common in the Sicilian population [62], are typically associated with a milder FA phenotype. Conversely, the FANCC c.456+4A>T PV is associated with a severe phenotype in the Ashkenazi Jewish population [69], yet this severity is not necessarily seen in Japanese individuals with the same PV [72].
- Population-specific carrier frequency:
- While the FA carrier frequency in the United States general population is estimated to be approximately 1:181 based on the reported incidence of FA, carrier frequency is higher in certain populations, such as Spanish Gypsies, Afrikaners, and Ashkenazi Jews, because of known founder events [1]. This information can inform reproductive counseling of individuals with a personal or family history of FA when their partner’s ethnic background places the partner at increased risk of being an FA carrier.
A summary of FANC gene founder variants is shown in Table 3.
Table 3. Founder pathogenic variants in FANC genes according to ethnic group.
| Gene | Pathogenic Variant(s) | Ethnicity | Carrier Frequency | Reference(s) |
|---|---|---|---|---|
| FANCA | c.37588_3790del | Brazilian | Unknown | [60] |
| c.295C>T | Spanish gypsy | 1 in 70 persons | [117] | |
| c.1007-?_3066+?del | Afrikaan | 1 in 80 persons | [118, 119] | |
| c.3398delA | Afrikaan | Unknown | [118] | |
| c.2546delC | Korean, Japanese | Unknown | [120, 121] | |
| c.3720_3724del | Korean, Japanese | Unknown | [120, 121] | |
| Deletion of exon 15 | Tunisian | Unknown | [122, 123] | |
| c.2222+166G>A | Tunisian | Unknown | [123] | |
| Deletions of exons 4 and 5 | Tunisian | Unknown | [123] | |
| 2172_2173insG | Moroccan | Unknown | [124] | |
| c.4275delT | Moroccan | Unknown | [124] | |
| c.890_893del | Tunisian | Unknown | [124] | |
| c.2574C>G | Indian | Unknown | [124] | |
| FANCC | c.456+4A>T | Ashkenazi Jewish | 1 in 90 persons | [125, 126] |
| c.165+1G>T | Saudi | Unknown | [74] | |
| c.456+4A>T | Japanese | Unknown | [72] | |
| c.67delG | Mennonite | Unknown | [73, 127] | |
| FANCD1/BRCA2 | c.8756delG | Cypriot | Unknown | [128, 129] |
| c.6174delT | Ashkenazi Jewish | 1-2 in 100 persons | [130] | |
| FANCD2 | c.1948-16T>G | Turkish | Unknown | [131] |
| FANCG/XRCC9 | c.637_643del | South African | 1 in 100 persons | [132] |
| c.307+1G>C | Korean, Japanese | Unknown | [120, 133] | |
| c.1066C>T | Korean, Japanese | Unknown | [120, 133] | |
| c.1480+1G>C | French Acadian | Unknown | [134] | |
| c.511-3_511-2delCA | Mixe (Mexico) | Unknown | [135] | |
| c.313G>T | German | Unknown | [136] | |
| c.1066C>T | Brazilian, Portuguese | Unknown | [134] | |
| c.1589_1591delATA | Korean | Unknown | [137] | |
| c.1649delC | Turkish | Unknown | [136] | |
| FANCL | c.1092G>A | South Asian | Unknown | [138] |
Sources for Table 3: [60, 72, 73, 74, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138]
Genetic Testing of Family Members
Once an individual’s genotype is known, family members can then undergo “targeted” analysis (also called carrier or single-site testing) to determine their carrier status and inform family planning. When possible, the affected individual should be tested first. However, if that person is unavailable for dedicated FA testing, panel testing to identify carrier status of relatives is a reasonable approach, although interpretation of the results may be complicated by VUS. Negative test results in an unaffected relative should also be interpreted with caution.
Owing to the clinical variability of FA (even within the same family), all biologic siblings of an individual with FA should undergo a chromosome breakage test. This is particularly important in the setting of transplantation when a family member is identified as a potential organ donor.
Inheritance of Fanconi Anemia
Fanconi anemia is predominantly inherited in an autosomal recessive manner, meaning that affected individuals harbor a disease-causing PV in both alleles of the same FANC gene. However, two forms deviate from this, with one following an X-linked inheritance pattern (FANCB) and the other being inherited in an autosomal dominant manner (FANCR/RAD51).
Autosomal Recessive Inheritance
In an autosomal recessive pattern of inheritance, an individual must have a PV on both copies of the same FANC gene (biallelic alteration) to have the condition. Biologic parents of an individual with FA should be offered carrier testing to confirm that each parent carries one of the known PVs and that the variants are in trans (on separate gene copies). Although rare, it is possible that a parent tests negative. Explanations for this negative result include the following:
- The egg or sperm involved in the child’s conception developed a spontaneous change (known as a de novo variant).
- Only a fraction of the parent’s reproductive cells contains the variant (known as germline or gonadal mosaicism).
- Uniparental disomy occurred, in which one variant was present on both gene copies and was inherited from only one parent. This has only been reported to date in patients with FANCA and FANCP mutations [139]
- The child was adopted, was the product of a donor egg or sperm, or the paternity or maternity was not accurately reported.
- Samples were mixed up during the testing process.
With autosomal recessive inheritance, when both parents are confirmed to be carriers of the PV, each child has a 25% chance of having FA. Unaffected siblings (with a negative chromosome breakage test) have a 67% chance of being a carrier of the FA gene PV.
Inheritance is also an important consideration when an individual with FA reaches reproductive age. While reduced fertility has been reported, some individuals have had biologic children [140, 141]. The likelihood of having an affected child depends on the genetic status of the partner. It is recommended that the partner undergo sequencing and deletion and/or duplication analysis of the causative FANC gene. For example, when FA is due to the FANCA PV in an affected individual, comprehensive FANCA analysis of their partner is indicated. Depending on a couple’s genetic status, pregnancy outcomes are as follows:
- If the partner tests negative, the likelihood of having a child with FA is very low. All children will be carriers.
- If the partner tests positive for a PV in the same FANC gene, there is a 50% chance that each child will have FA and a 50% likelihood that each child will be a FA carrier.
- If both partners have FA because of a PV in the same FANC gene, all (100%) of their children will have FA.
- If both partners have FA because of variants in different FANC genes, their children will be carriers for two different forms of FA (double heterozygous). The likelihood of their children being affected is very low, presuming that comprehensive testing for the FANC gene associated with the other partner’s FA diagnosis is negative in both individuals.
When an autosomal recessive form of FA is identified in a family, extended relatives on both sides of the family can be offered carrier testing for the known familial FANC variants.
X-Linked Inheritance
With an X-linked condition, the disease-causing gene resides on the X chromosome. In FA, this inheritance applies to the FANCB gene. Women have two X chromosomes, while men have one X and one Y chromosome. If a woman carries one FANCB PV (heterozygous), there is a 50% chance of passing the PV to each child. Any sons who inherit the PV will be affected. Any daughters who inherit the PV will be carriers. If carrier testing of the mother of an affected boy is negative, the boy’s FA PV likely appeared de novo, although germline mosaicism cannot be excluded. As such, male siblings of a patient with FA secondary to an FANCB PV should undergo chromosome breakage testing or targeted genetic testing for the familial PV. Maternal relatives in these families have an increased likelihood of being a carrier or having the condition. Any daughters born to affected men will be obligate carriers, whereas any sons will be unaffected because they inherit a Y chromosome from their fathers.
Autosomal Dominant Inheritance
Autosomal dominant inheritance means that only one copy of an abnormal gene is required to exhibit the condition. In FA, an autosomal dominant pattern of inheritance is seen only with a PV of the applies to the FANCR/RAD51 gene. While all affected individuals reported to date have had a de novo PV (i.e., the condition was not inherited from an affected parent), the parents should still be offered testing. Even if this testing is negative, gonadal (germline) mosaicism cannot be excluded; thus, there is still a small chance that siblings or a future pregnancy could be affected. Therefore, all siblings should undergo chromosome breakage testing, and anyone who tests positive is considered to have FA. Any child of an individual with FANCR/RAD51-associated FA will have a 50% chance of having FA and a 50% chance of not having FA.
Carrier Cancer Risk
Fanconi anemia and hereditary cancer genes encode proteins that function within a common pathway, called the FA DNA repair pathway. These proteins act together to maintain genome integrity by repairing DNA interstrand crosslinks, which is a type of DNA damage (see Chapter 1) [7, 142]. Cancer risk estimates for carriers (heterozygous) of a single PV in an FANC gene are rapidly evolving as new information becomes available [7]. The National Comprehensive Cancer Network (NCCN) reviews new literature regularly and provides updated clinical practice guidelines for the detection, prevention, and risk reduction of cancers linked to these genes [143]. It is important to note that panel tests from some laboratories may include FANC genes (in particular, moderate-risk genes) with insufficient data to fully determine the magnitude of their risks and offer management strategies [144, 145, 146].
For individuals who test positive for PVs in one of the FANC genes listed in Table 4 (at least at the time of this publishing) there is some degree of cancer risk that is observed by the NCCN.
Table 4. Cancer risks in carriers that may potentially affect management.
| FANC Gene | Established Cancer Risk in Carriers | Additional References |
|---|---|---|
| FANCD1/BRCA2* | Breast, ovary, prostate, pancreas, melanoma | 143, 147, 148] |
| FANCJ/BRIP1/BACH1 | Ovary | [146, 149, 150] |
| FANCN/PALB2 | Breast, pancreas, ovary | 146, 147, 150-157] |
| FANCO/RAD51C | Ovary, breast | [150, 158-163] |
| FANCS/BRCA1* | Breast, ovary, prostate, pancreas | [143, 147, 150, 164] |
*Hypomorphic variants noted.
Sources for Table 4: [143, 146, 147, 148, 149, 150, 151, 152, 153, 154, 155, 156, 157, 158, 159, 160, 161, 162, 163, 164]
The possibility of increased cancer risk in carriers of other FANC genes is a topic of much interest and ongoing research. Over the years, researchers have used data from FA families to compare the observed number of cancers in carriers of other FANC PVs with the estimated incidence of cancer in the general population. These studies, which were limited in scale and focused on self-reported data collection, did not reveal a significant difference in overall cancer risk for these carriers [165, 166, 167, 168, 169].
All FA carriers should be encouraged to communicate their genetic status with their primary providers and to follow-up annually for potential health and cancer risk updates. Referral to a genetics counselor who specializes in cancer predisposition is recommended for accurate risk assessment and a comprehensive discussion about testing, management options, and family planning.
Reproductive Planning
There are multiple reproductive options for parents of a child with FA and for individuals with FA. Preconception genetic counseling is available for families to discuss these options in greater detail.
Prenatal Diagnostic Testing
If there is concern for Fanconi anemia (FA) in the pregnancy due to either a family history or ultrasound findings, prenatal diagnostic testing can be performed to determine whether the fetus has FA. Prenatal testing may also be used to determine whether the fetus has the same human leukocyte antigens (HLA) as a sibling with FA. This process, known as HLA typing, will indicate whether the child is a suitable donor for the sibling with FA, and if so if cord blood banking might be beneficial. Prenatal testing options currently involve chorionic villus sampling (CVS) or amniocentesis procedures. CVS is typically performed between 10-14 weeks gestation and amniocentesis may be performed after the 15th week gestation. The goal of both procedures is to obtain fetal cells for genetic testing and/or chromosomal breakage analysis. If the pathogenic variants are known, targeted variant analysis should be performed on these samples whereas chromosome breakage analysis can be performed when the pathogenic variants are unknown, and FA is suspected based on ultrasound findings. Newer testing that is available at some centers includes genetic testing by multi-gene panels, whole exome or whole genome sequencing. These may be offered in the absence of known familial disease-causing variants when a diagnosis is suspected in the pregnancy. Both CVS and amniocentesis procedures are invasive, though they are associated with a less than 1% risk of miscarriage which should be discussed in detail with the center performing the procedure [170, 171]. It is important to discuss the various considerations and complexities of this testing with a prenatal genetic counselor and experienced OB/Gyn.
Prenatal diagnosis for FA enables improved education and preparation of the patient family and medical team. This includes enabling a patient family the opportunity to make reproductive decisions including pregnancy continuation or termination where available. Further, it enables closer pregnancy monitoring, establishment of care with an appropriate medical care team and/or potential for interventions if available. In addition, there may be the possibility of a fetal therapy in the future (contact Dr. Czechowicz at Stanford Children’s to inquire). However, some families may not want a prenatal diagnosis especially if they are concerned regarding risks of the procedure or if this may not change their actions. These are very personal choices and families should be supported through these decisions. If prenatal diagnosis is not elected but there is concern for FA, newborn testing can be obtained through testing cord blood or peripheral blood via chromosome breakage analysis and/or genetic testing.
Prenatal Care
If a prenatal diagnosis confirms FA, it is important that the family receive thorough genetic counseling and discuss these finding with an experienced genetic counselor, maternal fetal medicine (MFM) specialist and FA physician. FA disease can be quite variable and close pregnancy monitoring is important to assess how FA may be impacting the fetus. More frequent fetal ultrasounds, echo and/or MRI may be considered. Findings from this screening can then guide recommendations on pregnancy management and delivery plans. If there are high risk features noted on prenatal screening, then delivering in a hospital with a high acuity NICU may be very important. Cord blood banking can also be considered though this is most beneficial in the setting of a healthy fetus that could serve as a donor for a sibling with FA. In the setting of a fetus with FA, no options currently exist for genetic correction of cord blood and most cord blood samples are used for research testing. Postnatal care should be guided by a dedicated pediatrician in collaboration with an experienced hematologist. It is also recommended to establish care at a center with FA expertise. General recommendations include assessment of the impact of FA disease, education on FA and available treatment options, blood count monitoring and first bone marrow evaluation at 2 years of age (see other chapters of guidelines).
In Vitro Fertilization with Preimplantation Genetic Testing
In vitro fertilization (IVF) is a reproductive technology that involves fertilization of a mature egg (ovum) with sperm in a laboratory to obtain an embryo. The embryo is then implanted to initiate pregnancy. This technology is often used by individuals and couples experiencing infertility; however, the technology can also be paired with a second technology, preimplantation genetic testing (PGT) for monogenic disorders to test embryos produced through IVF for familial genetic disorder(s). While PGT is used in an attempt to select embryos without FA and those that are an HLA match for an affected sibling, this technology is not always successful in selecting embryos meeting these criteria. PGT centers should inform families of the experience and accuracy of their center.
Parents considering PGT should be advised of the chances of selecting a healthy, HLA-matched embryo. Theoretically, for couples with a child with an autosomal recessive form of FA, there is a 75% chance that an embryo will not have FA and a 25% chance that an embryo will be an HLA match; thus, the odds of an embryo being both unaffected with FA and an HLA match is 18.75% (3/16). Realistically, many couples require multiple rounds of IVF-PGT to achieve a clinical pregnancy resulting in a live birth. It is also recommended that prenatal testing be performed for all pregnancies resulting from embryos produced through IVF-PGT to confirm the expected genetic status.
Other Reproductive Options
Other reproductive options include the use of donor gametes (egg or sperm), adoption, and unassisted pregnancy. Surrogacy is also an option, especially for women with FA who have fertility issues or are concerned about the health implications of pregnancy.
Summary
Close communication between physicians, genetics counselors, cytogenetics laboratories, and molecular genetics laboratories is critical for establishing the diagnosis of FA and providing optimal care for patients with the disease. Early diagnosis of FA and characterization of patient-specific FA pathogenic variants are of utmost importance, as this information may influence the management and follow-up of patients and their families. It is imperative that a clinically certified laboratory perform the diagnostic tests to ensure adherence to rigorous standards for quality control and quality assurance. All cytogenetic findings should be interpreted within the context of the patient’s complete hematologic profile and other clinical features to obtain a comprehensive assessment of the patient’s status. It is recommended that a genetics counselor or other genetics professional help guide diagnostic genetic testing. A comprehensive review of the genetic findings, including the phenotype and cancer risks associated with certain FANC genes and the health and reproductive implications for other relatives, should be shared with the patient and family. Family counseling should include a review of the various reproductive options available for families who already have a child with FA, as well as for individuals with FA who wish to have children.
The Fanconi Cancer Foundation recognizes the following author contributions:
Evaluation for diagnosis and genotype-phenotype association sections:
Jeffrey Lipton, MD, PhD
Moisés Fiesco-Roa, MD, MSc, PhD(c)
Diagnostic and somatic testing sections:
Susan Olson, PhD, FACMG
Lisa Moreau, MS
Akiko Shimamura, MD, PhD
Genetic counseling, genetic variants, and diagnostic testing sections:
Rebecca Tryon, MS, MA, CGC*
Jennifer Kennedy, MS, CGC
Prenatal section:
Agnieszka Czechowicz, MD, PhD
Edited by Shelby Sharp, PhD
*Section Committee Chair
References
- 1. Rosenberg et al., How high are carrier frequencies of rare recessive syndromes? Contemporary estimates
- for Fanconi Anemia in the United States and Israel. Am J Med Genet A, 2011. 155A(8): p. 1877-1883.
- 2. Fiesco-Roa et al., Dysmorphology as a clinical tool for an early diagnosis of Fanconi Anemia. Acta Pediatr Mex, 2022. 43(2): p. 129-410.
- 3. Parikh and Bessler, Recent insights into inherited bone marrow failure syndromes. Curr Opin Pediatr, 2012. 24(1): p. 23-32.
- 4. Shimamura and Alter, Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev, 2010. 24(3): p. 101-122.
- 5. Svahn et al., Somatic, hematologic phenotype, long-term outcome, and effect of hematopoietic stem cell transplantation. An analysis of 97 Fanconi anemia patients from the Italian national database on behalf of the Marrow Failure Study Group of the AIEOP (Italian Association of Pediatric Hematology-Oncology). Am J Hematol, 2016. 91(7): p. 666-671.
- 6. Auerbach, Fanconi anemia and its diagnosis. Mutat Res, 2009. 668(1-2): p. 4-10.
- 7. Fiesco-Roa et al., Fanconi anemia and dyskeratosis congenita/telomere biology disorders: Two inherited bone marrow failure syndromes with genomic instability. Frontiers in Oncology, 2022.12: p. 949435.
- 8. Solomon et al., Clinical geneticists' views of VACTERL/VATER association. Am J Med Genet A, 2012. 158A(12): p. 3087-3100.
- 9. Fiesco-Roa et al., Genotype-phenotype associations in Fanconi anemia: A literature review. Blood Rev, 2019: p. 100589.
- 10. Alter and Giri, Thinking of VACTERL-H? Rule out Fanconi Anemia according to PHENOS. Am J Med Genet A, 2016. 170(6): p. 1520-1524.
- 11. Faivre et al., Should chromosome breakage studies be performed in patients with VACTERL association? Am J Med Genet A, 2005. 137(1): p. 55-58.
- 12. Risitano et al., Twenty years of the Italian Fanconi Anemia Registry: where we stand and what remains to be learned. Haematologica, 2016. 101(3): p. 319-327.
- 13. Mahmood et al., An experience with 124 cases of fanconi anemia: clinical spectrum, hematological parameters and chromosomal breakage analysis. Am J Blood Res, 2021. 11(5): p. 498-503.
- 14. Mori et al., Pathogenic mutations identified by a multimodality approach in 117 Japanese Fanconi anemia patients. Haematologica, 2019. 104(10): p. 1962-1973.
- 15. De Kerviler et al., The clinical and radiological features of Fanconi's anaemia. Clin Radiol, 2000. 55(5): p. 340-345.
- 16. Aksu et al., Central nervous system lesions in Fanconi anemia: Experience from a research center for Fanconi anemia patients. Pediatr Blood Cancer, 2020. 67(12): p. e28722.
- 17. George et al., A comprehensive molecular study identified 12 complementation groups with 56 novel FANC gene variants in Indian Fanconi anemia subjects. Hum Mutat, 2021. 42(12): p. 1648-1665.
- 18. Johnson-Tesch et al., Fanconi anemia: correlating central nervous system malformations and genetic complementation groups. Pediatr Radiol, 2017. 47(7): p. 868-876.
- 19. Stivaros et al., Central nervous system abnormalities in Fanconi anaemia: patterns and frequency on magnetic resonance imaging. Br J Radiol, 2015. 88(1056): p. 20150088.
- 20. Tsilou et al., Ocular and orbital manifestations of the inherited bone marrow failure syndromes: Fanconi anemia and dyskeratosis congenita. Ophthalmology, 2010. 117(3): p. 615-622.
- 21. Richards et al., Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 2015. 17(5): p. 405-424.
- 22. Auerbach, Fanconi anemia diagnosis and the diepoxybutane (DEB) test. Exp Hematol, 1993. 21(6): p. 731-733.
- 23. Auerbach, Diagnosis of Fanconi anemia by diepoxybutane analysis. Curr Protoc Hum Genet, 2015. 85: p. 8.7.1-8.7.17.
- 24. Castella et al., Chromosome fragility in patients with Fanconi anaemia: diagnostic implications and clinical impact. J Med Genet, 2011. 48(4): p. 242-250.
- 25. ACMG. Practice Guidelines. 2020; Available from: https://www.acmg.net/ACMG/Medical-Genetics-Practice-Resources/Practice-Guidelines.aspx.
- 26. Hirsch et al., Association of biallelic BRCA2/FANCD1 mutations with spontaneous chromosomal instability and solid tumors of childhood. Blood, 2004. 103(7): p. 2554-2559.
- 27. Chrzanowska, K.H., et al., Nijmegen breakage syndrome (NBS). Orphanet J Rare Dis, 2012. 7: p. 13.
- 28. Martin et al., Mutations in TOP3A Cause a Bloom Syndrome-like Disorder. Am J Hum Genet, 2018. 103(2): p. 221-231.
- 29. McKay et al., A Roberts Syndrome Individual With Differential Genotoxin Sensitivity and a DNA Damage Response Defect. Int J Radiat Oncol Biol Phys, 2019. 103(5): p. 1194-1202.
- 30. Terabayashi and Hanada, Genome instability syndromes caused by impaired DNA repair and aberrant DNA damage responses. Cell Biol Toxicol, 2018. 34(5): p. 337-350.
- 31. van der Lelij et al., Diagnostic Overlap between Fanconi Anemia and the Cohesinopathies: Roberts Syndrome and Warsaw Breakage Syndrome. Anemia, 2010. 2010: p. 565268.
- 32. Wu, Phenotypes and genotypes of the chromosomal instability syndromes. Transl Pediatr, 2016. 5(2): p. 79-83.
- 33. Aguilar-Martinez et al., Cytogenetic abnormalities in dyskeratosis congenita--report of five cases. Clin Exp Dermatol, 1988. 13(2): p. 100-104.
- 34. Molina et al., Fanconi anemia, Part 1. Cytogenetic diagnosis. Acta Pediatr Mex, 2022. 43(2): p. 102-128.
- 35. Auerbach et al., Prenatal and postnatal diagnosis and carrier detection of Fanconi anemia by a cytogenetic method. Pediatrics, 1981. 67(1): p. 128-135.
- 36. Cervenka and Hirsch, Cytogenetic differentiation of Fanconi anemia, "idiopathic" aplastic anemia, and Fanconi anemia heterozygotes. Am J Med Genet, 1983. 15(2): p. 211-223.
- 37. Technologists., A.o.C., The AGT cytogenetics laboratory manual. 3rd ed. / ed, ed. M.J. Barch, T. Knutsen, and J.L. Spurbeck. 1997, Philadelphia: Lippincott-Raven Publishers.
- 38. Nicoletti et al., Mosaicism in Fanconi anemia: concise review and evaluation of published cases with focus on clinical course of blood count normalization. Ann Hematol, 2020. 99(5): p. 913-924.
- 39. Gille et al., Diagnosis of Fanconi Anemia: Mutation Analysis by Multiplex Ligation-Dependent Probe Amplification and PCR-Based Sanger Sequencing. Anemia, 2012. 2012: p. 603253.
- 40. Knies et al., Genotyping of fanconi anemia patients by whole exome sequencing: advantages and challenges. PLoS One, 2012. 7(12): p. e52648.
- 41. Cheung and Taniguchi, Recent insights into the molecular basis of Fanconi anemia: genes, modifiers, and drivers. Int J Hematol, 2017. 106(3): p. 335-344.
- 42. Tsui and Crismani, The Fanconi Anemia Pathway and Fertility. Trends Genet, 2019. 35(3): p. 199-214.
- 43. Chandrasekharappa et al., Massively parallel sequencing, aCGH, and RNA-Seq technologies provide a comprehensive molecular diagnosis of Fanconi anemia. Blood, 2013. 121(22): p. e138-148.
- 44. Flynn et al., Comprehensive Analysis of Pathogenic Deletion Variants in Fanconi Anemia Genes. 2014. 35(11): p. 1342-1353.
- 45. Winberg et al., Mutation screening and array comparative genomic hybridization using a 180K oligonucleotide array in VACTERL association. PLoS One, 2014. 9(1): p. e85313.
- 46. Li et al., Functional analysis of Fanconi anemia mutations in China. Exp Hematol, 2018. 66: p. 32-41 e8.
- 47. Pilonetto et al., A strategy for molecular diagnostics of Fanconi anemia in Brazilian patients. Mol Genet Genomic Med, 2017. 5(4): p. 360-372.
- 48. Xue et al., Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: single-gene, gene panel, or exome/genome sequencing. Genet Med, 2015. 17(6): p. 444-451.
- 49. (OHRP), O.f.H.R.P. Genetic Information Nondiscrimination Act (GINA): OHRP Guidance. 2009 March 24, 2009; Available from: https://www.hhs.gov/ohrp/regulations-and-policy/guidance/guidance-on-genetic-information-nondiscrimination-act/index.html.
- 50. University, J.H., Online Mendelian Inheritance in Man, OMIM®. MIM: #612555. https://www.omim.org/entry/612555.
- 51. Kalia et al., Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genetics In Medicine, 2016. 19: p. 249.
- 52. Gregory et al., Somatic mosaicism in Fanconi anemia: evidence of genotypic reversion in lymphohematopoietic stem cells. Proc Natl Acad Sci U S A, 2001. 98(5): p. 2532-2537.
- 53. Ikeda et al., Genetic reversion in an acute myelogenous leukemia cell line from a Fanconi anemia patient with biallelic mutations in BRCA2. Cancer Res, 2003. 63(10): p. 2688-2694.
- 54. Cioc et al., Diagnosis of myelodysplastic syndrome among a cohort of 119 patients with fanconi anemia: morphologic and cytogenetic characteristics. Am J Clin Pathol, 2010. 133(1): p. 92-100.
- 55. Mehta et al., Numerical chromosomal changes and risk of development of myelodysplastic syndrome--acute myeloid leukemia in patients with Fanconi anemia. Cancer Genet Cytogenet, 2010. 203(2): p. 180-186.
- 56. Meyer et al., Chromosomal aberrations associated with clonal evolution and leukemic transformation in fanconi anemia: clinical and biological implications. Anemia, 2012. 2012: p. 349837.
- 57. Rochowski et al., Patients with Fanconi anemia and AML have different cytogenetic clones than de novo cases of AML. Pediatr Blood Cancer, 2012. 59(5): p. 922-924.
- 58. Tonnies et al., Clonal chromosomal aberrations in bone marrow cells of Fanconi anemia patients: gains of the chromosomal segment 3q26q29 as an adverse risk factor. Blood, 2003. 101(10): p. 3872-3874.
- 59. Faivre et al., Association of complementation group and mutation type with clinical outcome in fanconi anemia. European Fanconi Anemia Research Group. Blood, 2000. 96(13): p. 4064-4070.
- 60. Castella et al., Origin, functional role, and clinical impact of Fanconi anemia FANCA mutations. Blood, 2011. 117(14): p. 3759-3769.
- 61. Steinberg-Shemer et al., Characterization and genotype-phenotype correlation of patients with Fanconi anemia in a multi-ethnic population. Haematologica, 2020. 105(7): p. 1825-1834.
- 62. Bottega et al., Hypomorphic FANCA mutations correlate with mild mitochondrial and clinical phenotype in Fanconi anemia. Haematologica, 2018. 103(3): p. 417-426.
- 63. McCauley et al., X-linked VACTERL with hydrocephalus syndrome: further delineation of the phenotype caused by FANCB mutations. Am J Med Genet A, 2011. 155A(10): p. 2370-2380.
- 64. Mikat et al., X-linked recessive VACTERL-H due to a mutation in FANCB in a preterm boy. Clin Dysmorphol, 2016. 25(2): p. 73-76.
- 65. Asur et al., Somatic mosaicism of an intragenic FANCB duplication in both fibroblast and peripheral blood cells observed in a Fanconi anemia patient leads to milder phenotype. Mol Genet Genomic Med, 2018. 6(1): p. 77-91.
- 66. Jung et al., Clinical severity in Fanconi anemia correlates with residual function of FANCB missense variants. bioRxiv, 2019. 772574.
- 67. Kutler et al., A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood, 2003. 101(4): p. 1249-1256.
- 68. Gillio et al., Phenotypic consequences of mutations in the Fanconi anemia FAC gene: an International Fanconi Anemia Registry study. Blood, 1997. 90(1): p. 105-110.
- 69. Yamashita et al., Clinical variability of Fanconi anemia (type C) results from expression of an amino terminal truncated Fanconi anemia complementation group C polypeptide with partial activity. Blood, 1996. 87(10): p. 4424-4432.
- 70. Aftab et al., Analysis of FANCC gene mutations (IVS4+4A>T, del322G, and R548X)in patients with Fanconi anemia in Pakistan. Turk J Med Sci, 2017. 47(2): p. 391-398.
- 71. Rodriguez et al., Molecular analysis of the most prevalent mutations of the FANCA and FANCC genes in Brazilian patients with Fanconi anaemia. Genetics and Molecular Biology, 2005. 28: p. 205-209.
- 72. Futaki et al., The IVS4 + 4 A to T mutation of the fanconi anemia gene FANCC is not associated with a severe phenotype in Japanese patients. Blood, 2000. 95(4): p. 1493-1498.
- 73. de Vries et al., A Dutch Fanconi Anemia FANCC Founder Mutation in Canadian Manitoba Mennonites. Anemia, 2012. 2012: p. 865170.
- 74. Hartmann et al., Correct mRNA processing at a mutant TT splice donor in FANCC ameliorates the clinical phenotype in patients and is enhanced by delivery of suppressor U1 snRNAs. Am J Hum Genet, 2010. 87(4): p. 480-493.
- 75. Howlett et al., Biallelic inactivation of BRCA2 in Fanconi anemia. Science, 2002. 297.
- 76. Alter et al., Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J Med Genet, 2007. 44(1): p. 1-9.
- 77. Offit, BRCA mutation frequency and penetrance: new data, old debate. J Natl Cancer Inst, 2006. 98(23): p. 1675-1677.
- 78. Wagner et al., Germline mutations in BRCA2: shared genetic susceptibility to breast cancer, early onset leukemia, and Fanconi anemia. Blood, 2004. 103(8): p. 3226-3229.
- 79. McReynolds et al., Genotype-cancer association in patients with Fanconi anemia due to pathogenic variants in FANCD1 (BRCA2) or FANCN (PALB2). Cancer Genet, 2021. 258-259: p. 101-109.
- 80. Degrolard-Courcet et al., Development of primary early-onset colorectal cancers due to biallelic mutations of the FANCD1/BRCA2 gene. Eur J Hum Genet, 2014. 22(8): p. 979-987.
- 81. Rickman and Smogorzewska, Advances in understanding DNA processing and protection at stalled replication forks. J Cell Biol, 2019. 218(4): p. 1096-1107.
- 82. Weinberg-Shukron et al., Essential Role of BRCA2 in Ovarian Development and Function. N Engl J Med, 2018. 379(11): p. 1042-1049.
- 83. Ip et al., Catastrophic chemotherapy toxicity leading to diagnosis of Fanconi anaemia due to FANCD1/BRCA2 during adulthood: description of an emerging phenotype. J Med Genet, 2021. 59(9):912-915.
- 84. Caburet et al., Homozygous hypomorphic BRCA2 variant in primary ovarian insufficiency without cancer or Fanconi anaemia trait. J Med Genet, 2020.
- 85. Qin et al., BRCA2 in Ovarian Development and Function. N Engl J Med, 2019. 380(11): p. 1086.
- 86. Meetei et al., A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat Genet, 2005. 37(9): p. 958-963.
- 87. Singh et al., Impaired FANCD2 monoubiquitination and hypersensitivity to camptothecin uniquely characterize Fanconi anemia complementation group M. Blood, 2009. 114(1): p. 174-80.
- 88. Bogliolo et al., Biallelic truncating FANCM mutations cause early-onset cancer but not Fanconi anemia. Genet Med, 2018. 20(4): p. 458-463.
- 89. Catucci et al., Individuals with FANCM biallelic mutations do not develop Fanconi anemia, but show risk for breast cancer, chemotherapy toxicity and may display chromosome fragility. Genet Med, 2018. 20(4): p. 452-457.
- 90. Chang et al., Whole exome sequencing reveals concomitant mutations of multiple FA genes in individual Fanconi anemia patients. BMC Med Genomics, 2014. 7: p. 24.
- 91. Fouquet et al., A homozygous FANCM mutation underlies a familial case of non-syndromic primary ovarian insufficiency. Elife, 2017. 6.
- 92. Yin et al., A homozygous FANCM frameshift pathogenic variant causes male infertility. Genet Med, 2019. 21(1): p. 62-70.
- 93. Zhang et al., Novel Bi-Allelic Variants of FANCM Cause Sertoli Cell-Only Syndrome and Non-Obstructive Azoospermia. Front Genet, 2021. 12: p. 799886.
- 94. Encarnacion et al., Fanconi-like anemia related to a FANCM mutation. Eur J Med Genet, 2022. 65(1): p. 104399.
- 95. Reid, S., et al., Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet, 2007. 39(2): p. 162-164.
- 96. Ghazwani et al., Clinical characteristics and genetic subtypes of Fanconi anemia in Saudi patients. Cancer Genet, 2016. 209(4): p. 171-176.
- 97. Serra et al., Shared Copy Number Variation in Simultaneous Nephroblastoma and Neuroblastoma due to Fanconi Anemia. Mol Syndromol, 2012. 3(3): p. 120-130.
- 98. Byrd et al., A Hypomorphic PALB2 Allele Gives Rise to an Unusual Form of FA-N Associated with Lymphoid Tumour Development. PLoS Genet, 2016. 12(3): p. e1005945.
- 99. Jacquinet et al., Expanding the FANCO/RAD51C associated phenotype: Cleft lip and palate and lobar holoprosencephaly, two rare findings in Fanconi anemia. Eur J Med Genet, 2018. 61(5): p. 257-261.
- 100. Vaz et al., Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat Genet, 2010. 42(5): p. 406-409.
- 101. Ameziane et al., A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. Nat Commun, 2015. 6: p. 8829.
- 102. Wang et al., A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination. Mol Cell, 2015. 59(3): p. 478-490.
- 103. Depienne et al., RAD51 haploinsufficiency causes congenital mirror movements in humans. Am J Hum Genet, 2012. 90(2): p. 301-307.
- 104. Trouillard et al., Congenital Mirror Movements Due to RAD51: Cosegregation with a Nonsense Mutation in a Norwegian Pedigree and Review of the Literature. Tremor Other Hyperkinet Mov (N Y), 2016. 6: p. 424.
- 105. Demirayak et al., Abnormal subcortical activity in congenital mirror movement disorder with RAD51 mutation. Diagn Interv Radiol, 2018. 24(6): p. 392-401.
- 106. University, J.H., Online Mendelian Inheritance in Man, OMIM®. MIM: #614508. https://www.omim.org/entry/614508.
- 107. Bogliolo et al., Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet, 2013. 92(5): p. 800-806.
- 108. Kashiyama et al., Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am J Hum Genet, 2013. 92(5): p. 807-819.
- 109. Niedernhofer et al., A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature, 2006. 444(7122): p. 1038-1043.
- 110. Sijbers et al., Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell, 1996. 86(5): p. 811-822.
- 111. Domchek et al., Biallelic Deleterious <em>BRCA1</em> Mutations in a Woman with Early-Onset Ovarian Cancer. 2013. 3(4): p. 399-405.
- 112. Sawyer et al., Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov, 2015. 5(2): p. 135-142.
- 113. Seo et al., Mechanism for survival of homozygous nonsense mutations in the tumor suppressor gene <em>BRCA1</em>. 2018. 115(20): p. 5241-5246.
- 114. Freire et al., Homozygous loss of function BRCA1 variant causing a Fanconi-anemia-like phenotype, a clinical report and review of previous patients. Eur J Med Genet, 2018. 61(3): p. 130-133.
- 115. Chirita-Emandi et al., Biallelic variants in BRCA1 gene cause a recognisable phenotype within chromosomal instability syndromes reframed as BRCA1 deficiency. J Med Genet, 2021. 58(9): p. 648-652.
- 116. Keupp et al., Biallelic germline BRCA1 mutations in a patient with early onset breast cancer, mild Fanconi anemia-like phenotype, and no chromosome fragility. Mol Genet Genomic Med, 2019. 7(9): p. e863.
- 117. Callen et al., A common founder mutation in FANCA underlies the world's highest prevalence of Fanconi anemia in Gypsy families from Spain. Blood, 2005. 105(5): p. 1946-1949.
- 118. Tipping et al., Molecular and genealogical evidence for a founder effect in Fanconi anemia families of the Afrikaner population of South Africa. Proc Natl Acad Sci U S A, 2001. 98(10): p. 5734-5739.
- 119. Rosendorff et al., Fanconi anemia: another disease of unusually high prevalence in the Afrikaans population of South Africa. Am J Med Genet, 1987. 27(4): p. 793-797.
- 120. Park et al., FANCA and FANCG are the major Fanconi anemia genes in the Korean population. Clin Genet, 2013. 84(3): p. 271-275.
- 121. Yagasaki et al., Identification and characterization of novel mutations of the major Fanconi anemia gene FANCA in the Japanese population. Hum Mutat, 2004. 24(6): p. 481-490.
- 122. Amouri et al., High frequency of exon 15 deletion in the FANCA gene in Tunisian patients affected with Fanconi anemia disease: implication for diagnosis. Mol Genet Genomic Med, 2014. 2(2): p. 160-165.
- 123. Ben Haj Ali et al., FANCA Gene Mutations in North African Fanconi Anemia Patients. Front Genet, 2021. 12: p. 610050.
- 124. Tamary et al., Fanconi anaemia group A (FANCA) mutations in Israeli non-Ashkenazi Jewish patients. Br J Haematol, 2000. 111(1): p. 338-343.
- 125. Whitney et al., A common mutation in the FACC gene causes Fanconi anaemia in Ashkenazi Jews. Nat Genet, 1993. 4(2): p. 202-205.
- 126. Verlander et al., Carrier frequency of the IVS4 + 4 A-->T mutation of the Fanconi anemia gene FAC in the Ashkenazi Jewish population. Blood, 1995. 86(11): p. 4034-4038.
- 127. Garcia-de Teresa et al., FANCC Dutch founder mutation in a Mennonite family from Tamaulipas, Mexico. Mol Genet Genomic Med, 2019. 7(6): p. e710.
- 128. Hadjisavvas et al., Hereditary breast and ovarian cancer in Cyprus: identification of a founder BRCA2 mutation. Cancer Genet Cytogenet, 2004. 151(2): p. 152-156.
- 129. Loizidou et al., Fanconi anemia-D1 due to homozygosity for the BRCA2 gene Cypriot founder mutation: A case report. Oncol Lett, 2016. 11(1): p. 471-473.
- 130. Roa et al., Ashkenazi Jewish population frequencies for common mutations in BRCA1 and BRCA2. Nat Genet, 1996. 14(2): p. 185-187.
- 131. Kalb et al., Hypomorphic mutations in the gene encoding a key Fanconi anemia protein, FANCD2, sustain a significant group of FA-D2 patients with severe phenotype. Am J Hum Genet, 2007. 80(5): p. 895-910.
- 132. Morgan et al., A common Fanconi anemia mutation in black populations of sub-Saharan Africa. Blood, 2005. 105(9): p. 3542-3544.
- 133. Yagasaki et al., Two common founder mutations of the fanconi anemia group G gene FANCG/XRCC9 in the Japanese population. Hum Mutat, 2003. 21(5): p. 555.
- 134. Auerbach et al., Spectrum of sequence variation in the FANCG gene: an International Fanconi Anemia Registry (IFAR) study. Hum Mutat, 2003. 21(2): p. 158-168.
- 135. Reyes et al., Fanconi Anemia Patients from an Indigenous Community in Mexico Carry a New Founder Pathogenic Variant in FANCG. Int J Mol Sci, 2022. 23(4).
- 136. Demuth et al., Spectrum of mutations in the Fanconi anaemia group G gene, FANCG/XRCC9. Eur J Hum Genet, 2000. 8(11): p. 861-868.
- 137. Park et al., Founder haplotype analysis of Fanconi anemia in the Korean population finds common ancestral haplotypes for a FANCG variant. Ann Hum Genet, 2015. 79(3): p. 153-161.
- 138. Donovan et al., A founder variant in the South Asian population leads to a high prevalence of FANCL Fanconi anemia cases in India. Hum Mutat, 2020. 41(1): p. 122-128.
- 139. Donovan et al., Paternal or Maternal Uniparental Disomy of Chromosome 16 Resulting in Homozygosity of a Mutant Allele Causes Fanconi Anemia. Hum Mutat, 2016. 37(5): p. 465-468.
- 140. Alter et al., Fanconi's anaemia and pregnancy. Br J Haematol, 1991. 77(3): p. 410-418.
- 141. Nabhan et al., Fertility recovery and pregnancy after allogeneic hematopoietic stem cell transplantation in Fanconi anemia patients. Haematologica, 2010. 95(10): p. 1783-1787.
- 142. Kottemann and Smogorzewska, Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature, 2013. 493(7432): p. 356-363.
- 143. Gallagher et al., Germline BRCA mutations denote a clinicopathologic subset of prostate cancer. Clin Cancer Res, 2010. 16(7): p. 2115-2121.
- 144. Easton et al., Gene-panel sequencing and the prediction of breast-cancer risk. N Engl J Med, 2015. 372(23): p. 2243-2257.
- 145. Network, N.C.C. Genetic/Familial High-Risk Assessment: Breast and Ovarian. 2019 [cited 2019 11/20/2019]; Version 3.2010
- 146. Ramus et al., Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women With Ovarian Cancer. J Natl Cancer Inst, 2015. 107(11).
- 147. Norquist et al., Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol, 2016. 2(4): p. 482-490.
- 148. Tung et al., Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer. J Clin Oncol, 2016. 34(13): p. 1460-1468.
- 149. Rafnar et al., Mutations in BRIP1 confer high risk of ovarian cancer. Nat Genet, 2011. 43(11): p. 1104-1107.
- 150. Tung et al., Counselling framework for moderate-penetrance cancer-susceptibility mutations. Nat Rev Clin Oncol, 2016. 13(9): p. 581-588.
- 151. Antoniou et al., Breast-cancer risk in families with mutations in PALB2. N Engl J Med, 2014. 371(6): p. 497-506.
- 152. Couch et al., Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J Clin Oncol, 2015. 33(4): p. 304-311.
- 153. Grant et al., Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology, 2015. 148(3): p. 556-564.
- 154. Pritchard et al., Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N Engl J Med, 2016. 375(5): p. 443-453.
- 155. Lilyquist et al., Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi-gene panels compared to reference controls. Gynecol Oncol, 2017. 147(2): p. 375-380.
- 156. Kurian et al., Breast and Ovarian Cancer Penetrance Estimates Derived From Germline Multiple-Gene Sequencing Results in Women. JCO Precis Oncol, 2017. 1: p. 1-12.
- 157. Yang et al., Cancer Risks Associated With Germline PALB2 Pathogenic Variants: An International Study of 524 Families. J Clin Oncol, 2020. 38(7): p. 674-685.
- 158. Loveday et al., Germline RAD51C mutations confer susceptibility to ovarian cancer. Nat Genet, 2012. 44(5): p. 475-6; author reply 476.
- 159. Song et al., Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J Clin Oncol, 2015. 33(26): p. 2901-2907.
- 160. Breast Cancer Association et al., Breast Cancer Risk Genes - Association Analysis in More than 113,000 Women. N Engl J Med, 2021. 384(5): p. 428-439.
- 161. Hu et al., The Contribution of Germline Predisposition Gene Mutations to Clinical Subtypes of Invasive Breast Cancer From a Clinical Genetic Testing Cohort. J Natl Cancer Inst, 2020. 112(12): p. 1231-1241.
- 162. Li et al., Combined Tumor Sequencing and Case-Control Analyses of RAD51C in Breast Cancer. J Natl Cancer Inst, 2019. 111(12): p. 1332-1338.
- 163. Yang et al., Ovarian and Breast Cancer Risks Associated With Pathogenic Variants in RAD51C and RAD51D. J Natl Cancer Inst, 2020. 112(12): p. 1242-1250.
- 164. Leongamornlert et al., Germline BRCA1 mutations increase prostate cancer risk. Br J Cancer, 2012. 106(10): p. 1697-1701.
- 165. Alter et al., Cancer in Heterozygote Carriers of Fanconi Anemia Genes. Blood, 2018. 132: p. 3868-3868.
- 166. Berwick et al., Genetic heterogeneity among Fanconi anemia heterozygotes and risk of cancer. Cancer Res, 2007. 67(19): p. 9591-9596.
- 167. Swift et al., Reassessment of cancer predisposition of Fanconi anemia heterozygotes. J Natl Cancer Inst, 1980. 65(5): p. 863-867.
- 168. Tischkowitz et al., Cancer incidence in relatives of British Fanconi Anaemia patients. BMC Cancer, 2008. 8: p. 257.
- 169. McReynolds et al., Risk of cancer in heterozygous relatives of patients with Fanconi anemia. Genet Med, 2022. 24(1): p. 245-250.
- 170. Akolekar et al., Procedure-related risk of miscarriage following amniocentesis and chorionic villus sampling: a systematic review and meta-analysis. Ultrasound Obstet Gynecol, 2015. 45(1): p. 16-26.
- 171. Likar et al., Pregnancy Loss After Amniocentesis and Chorionic Villus Sampling: Cohort Study. Zdr Varst, 2020. 60(1): p. 25-29.